Термин «нейрокогнитивные расстройства» (НКР) обозначает группу состояний, которая соотносится с разделом DSM-IV (1994,2000) «Деменция, Делирий, Амнестическое и другие когнитивные расстройства», она включает делирий и синдромы большого и малого НКР с их этиологическими типами.
Этиология
Заболевания с преобладанием когнитивных нарушений имеют различную природу. Из состояний, приведённых в DSM-5 (2013), можно сформировать следующие группы причин:
- Нейродегенеративные заболевания: болезнь Альцгеймера, болезнь диффузных телец Леви, болезнь Гентингтона.
- Сосудистые заболевания: болезнь «мелких сосудов», в том числе церебральная амилоидная ангиопатия, последствия острых нарушений мозгового кровообращения и их сочетание.
- Последствия черепно-мозговых травм.
- Нейроинфекции, включая ВИЧ.
- Прионные заболевания.
- Другие заболевания (гипоксия, обменно-метаболические и иммунно-опосредованные нарушения).
- Действие нескольких причин.
- Неуточнённые причины.
Обратимые причины деменции
.jpg)
Патогенез наиболее распространённых форм деменции
Болезнь Альцгеймера
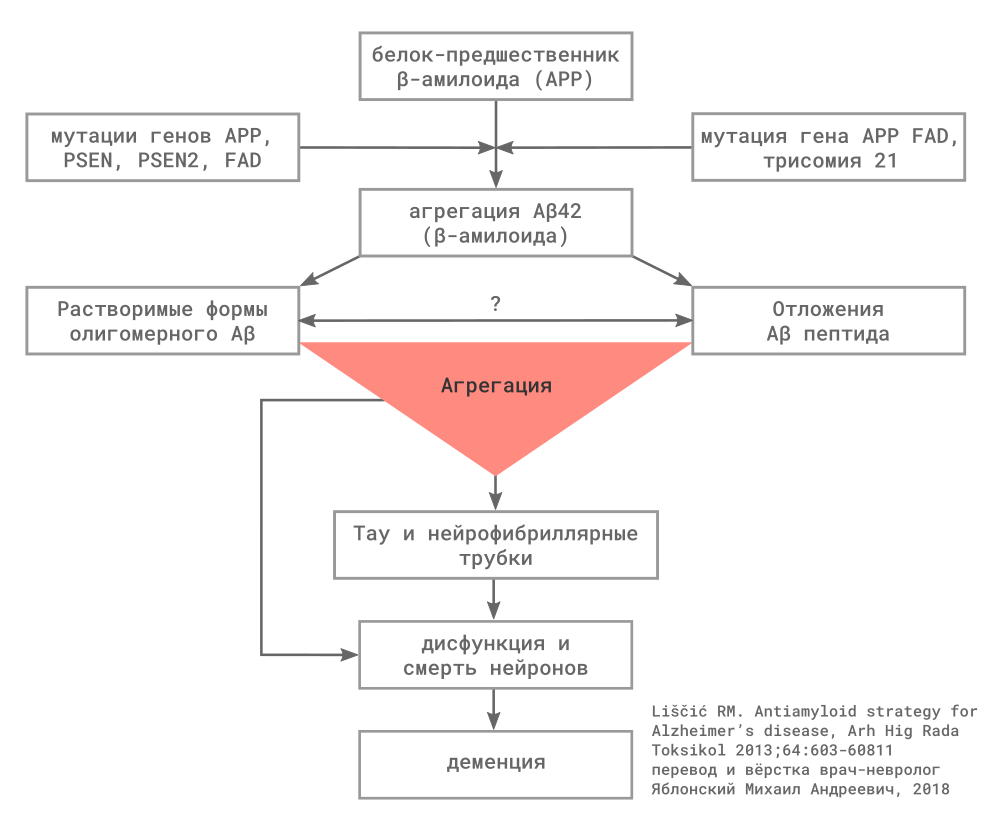
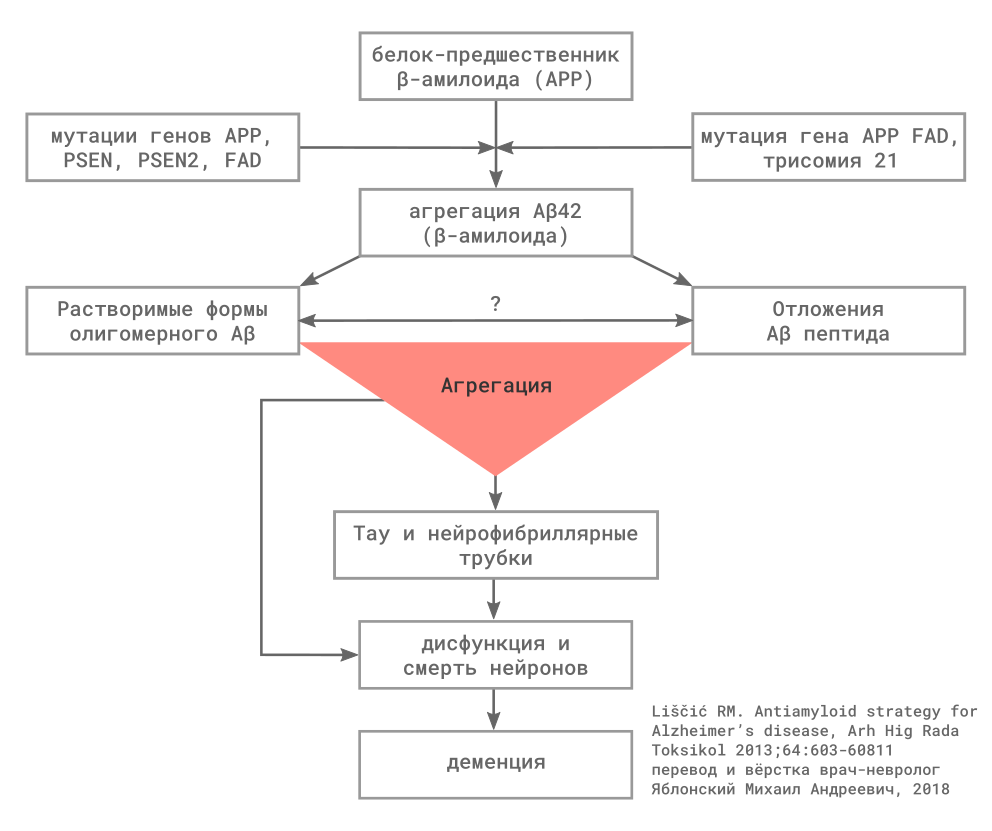
Для всех форм болезни Альцгеймера (БА) характерны повышенная продукция и/или замедленное разрушение амилоидных β — пептидов.
Главным патологическим признаком БА являются диффузные невритические бляшки (хорошо различимые скопления амилоида в межклеточном пространстве головного мозга) и нейрофибриллярные трубочки, внеклеточные агрегаты, включающие в состав гиперфосфорилированный тау-протеин (p-tau).
Амилоидные β — пептиды (в патогенезе БА наиболее значим Aβ42) образуются в межклеточном пространстве при расщеплении белка-предшественника амилоида (APP) под действием ферментов β и γ — секретаз. Пресенилин входит в состав комплекса с γ — секретазой, мутация пресенилина 1 (PSEN1) или пресенилин 2 (PSEN2), вероятно, способствует образованию амилоидных пептидов в-целом или образованию их более нейротоксичных форм. До настоящего времени не определено, какое вещество является основным нейротоксином, результаты экспериментальных исследований подчёркивают роль малых агрегатов амилоидных β — пептидов, которые называются «олигомеры», в противоположность более крупным агрегатам, которые называются «фибриллы».
Второй белок, включённый в патогенез БА, — тау. Тау, связываясь с микротрубочками, обеспечивает их стабильность. При БП, тау становится гиперфосфорилированным и агрегируется с образованием парных спиралевидных филаментов, основного компонента нейрофибриллярных трубочек в цитоплазме нейронов. Накопление этого изменённого белка оказывает токсическое действие на нейроны экспериментальных модели, которое предполагается в клинических условиях. Кроме того, предполагается, что передача патологических форм тау протеина между нейронами отвечает за распространение изменений альцгеймеровского типа в ткани мозга, вовлекая с прогрессированием заболевания всё новые области.
Существует ещё несколько важных и потенциально взаимодействующих механизмов, вероятно, включенных в патогенез БА, для многих из них выделены гены, повышающие риск развития заболевания. Например, белок аполипопротеин Е человека (ApoE) — липопротеин с широким набором свойств, принимающий участие во множественных клеточных процессах, включая транспорт холестерина, пластичность синапсов, регуляция иммунного ответа. Выделено три аллеля гена ApoE, которые обозначаются как эпсилон 2 (ε2), ε3, and ε4, которые кодируют изоформы с различной активностью. Установлен как минимум один механизм, который объясняет повышение риска БА у носителей аллеля ApoE ε4, нарушением клиренса амилоида в головном мозге.
В дополнение этим основным патологическим признакам, описан ряд изменений, которые часто встречаются у больных БА.
Церебральная амилоидная ангиопатия часто сочетается с отложением амилоида в паренхиме головного мозга.
Включения аномального альфа-синуклеина — тельца Леви, часто встречаются на промежуточных и распространённых стадиях патологических изменений больных БА. Наибольшее количество телец Леви часто выявляется в миндалине.
Сосудистые изменения ткани головного мозга (см. сосудистые НКР)
Склероз гиппокампа у больных БА определяется как утрата пирамидных клеток и глиоз. Также встречается при лобно-височной деменции, сосудистом поражении головного мозга, хронической эпилепсии.
Иммунореактивные включения транзактивного ДНК-связывающего белка 43 (TDP-43), как в большинстве случаев бокового амиотрофического склероза и ряде случаев лобно-височной деменции, часто встречаются при БА.
Есть данные о сочетании гиппокампальных отложений TDP-43 с более быстрым прогрессированием атрофии гиппокампа у больных БА.
Рисунок 1
Биохимические трансформации амилоида и их роль в патогенезе болезни Альцгеймера
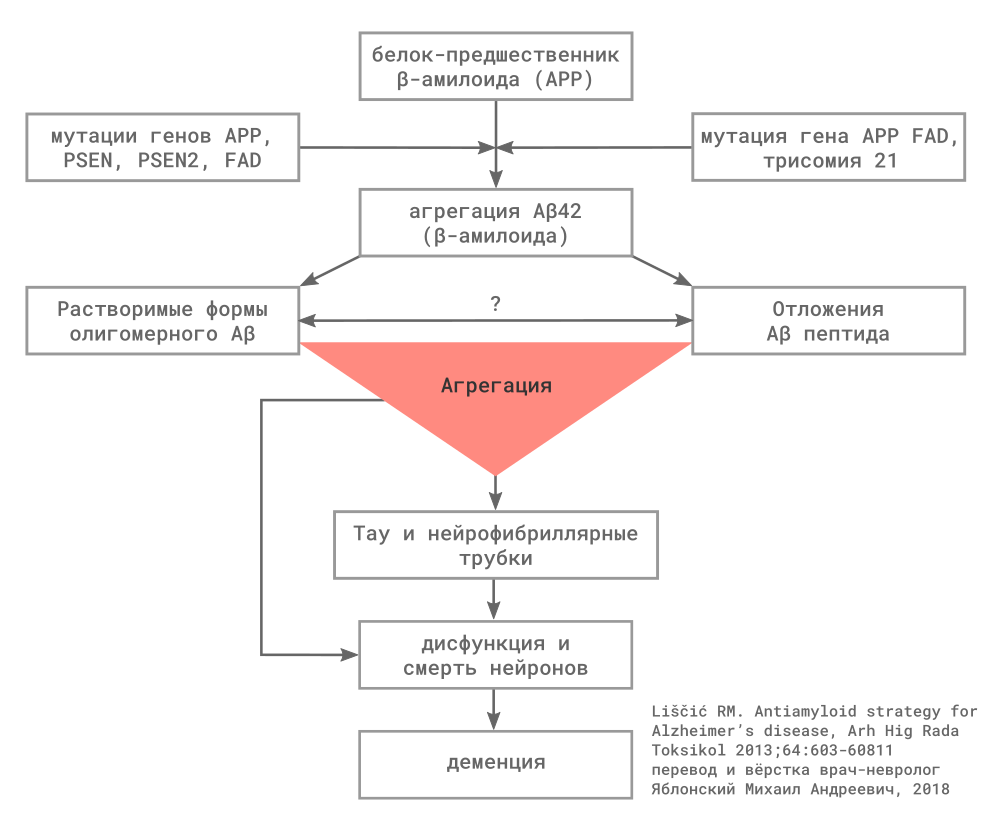
Рисунок 2
Механизм образования амилоидного β — пептида
Сосудистая деменция
Наиболее значимым фактором риска поражения мелких сосудов головного мозга является артериальная гипертензия. Другими значимыми повреждающими факторами являются сахарный диабет и курение. При цереброваскулярном заболевании встречаются как первичные сосудистые, так и вторичные дегенеративные изменения вещества головного мозга.
Выделяют несколько видов патологических изменений вещества головного мозга, ассоциированных с хроническим ишемическим повреждением.
- постинфарктные очаги (»неполные» и «немые» инфаркты),
- лейкоареоз (диффузные изменения белого вещества головного мозга),
- множественные лакуны,
- расширенные периваскулярные пространства,
- Последствия кровоизлияний,
- Атрофия вещества головного мозга,
Атеросклероз поражает артерии эластического и крупные артерии мышечного типа. В большинстве случаев атеросклероз приводит к развитию острых нарушений мозгового кровообращения с быстрым развитием выраженных очаговых симптомов. В патогенезе хронических сосудистых заболеваний головного мозга основное значение имеет поражение микроциркуляторного русла. Оно образовано терминальными ветвями крупных артерий каротидного и вертебрально-базилярного бассейна, а также притоками церебральных вен. Артерии микроциркуляторного русла подвержены изменениями при резких перепадах артериального давления поскольку на этом уровне не образуются анастомозы.
Болезнь мелких сосудов или микроангиопатия может иметь различной этиологии:
- например, липогиалиноз (артериосклероз) с развитием лакунарных инфарктов;
- церебральная аутосомная доминантная артериопатия с лакунарными инфарктами (CADASIL);
- митохондриальная энцефалопатия, лактат-ацидоз с инсультоподобными эпизодами (MELAS);
- Болезни накопления: накопление амилоида при церебральном амилоидозе;
- или связанную с неуточнённой причиной (предполагаются иммунологические, воспалительные изменения, интоксикация).
В ряде случаев, нарушения микроциркуляции могут быть вызваны патологией со стороны системы крови, без поражения кровеносных сосудов. Истинная полицитемия, гемоглобинопатии или протеинопатии могут повышать вязкость крови, что приводит к сопротивлению кровотоку и агрегации форменных элементов, является причиной церебральной ишемии. В редких случаях возможно развитие микроэмболии. Важными дополнительными факторами могут быть нарушения обмена веществ, прежде всего, сахарный диабет и нарушение венозного оттока.
Нарушения ауторегуляции мозгового кровообращения являются как причиной, так и следствием поражения микрососудов, принимая участие в порочном круге патологических изменений.
Лейкоареоз — нейровизуализационный термин (снижение рентгеновской плотности на КТ головного мозга, повышение интенсивности сигнала по данным МРТ в Т2/FLAIR режимах), описывающий неспецифические изменения подкоркового белого вещества головного мозга. Выявляются двусторонние очаговые, иногда сливные изменения. В основе лейкоареоза лежит поражение мелких артерий и артериол вследствие липогиалиноза (также обозначается как артериосклероз) и микроатероматоза.
Факторами риска развития лейкоареоза являются возраст, повышенное артериальное давление и сахарный диабет. Повышение внутрижелудочкового давления рассматривается в качестве дополнительного механизма развития лейкоареоза, оно приводит к нарушению микроциркуляции в прилегающих к желудочкам участках головного мозга. У пациентов с нормотензивной гидроцефалией отмечаются распространённые изменения подкоркового белого вещества.
Доказана связь распространенности диффузных изменений белого вещества головного мозга с выраженностью депрессии и когнитивных нарушений.
В препаратах головного мозга лейкоареоз часто сочетается с расширением периваскулярных (Вирхова-Робина) пространств — криблюрами (‘etát criblé’). Этот феномен связывают с повторными эпизодами ишемии и плазматического пропитывания участков ткани, окружающих микрососуды.
Липогиалиноз — дистрофический процесс, поражающий пенетрирующие артерии и артериолы диаметром до 200 мкм. Протекает под действием повышения артериального давления, гипергликемии, а также вследствие возрастных изменений. Повышение тканево-сосудистой проницаемости в субэндотелиальном пространстве сосуда приводит к поступлению туда белков плазмы и адсорбции их на изменённых волокнистых структурах соединительной ткани с последующей преципитацией и образованием белка гиалина. В последующем, гиалин оттесняет и разрушает эластичную мембрану, средняя оболочка истончается, и перфорирующая артерия приобретает вид стекловидной трубочки. Просвет артерии резко сужается, возможно развитие окклюзии.
При развитии липогиалиноза фоне сахарного диабета субэндотелиально часто откладывается не только гиалин, но и липиды. Сосудистая стенка инфильтрируется макрофагами с включениям фагоцитированных липидов — пенистыми клетками. В изменённых тканях происходит отложение фибриноида (сложного вещества, состоящего из белков и полисахаридов, распадающихся коллагеновых волокон, межклеточного вещества и плазмы крови). В неблагоприятных условиях происходит гибель окружающих тканей — фибриноидный некроз. Этот патологических процесс выявляется в артериолах и капиллярах головного мозга, сетчатки и почек часто сочетается с гиалинозом.
Вследствие нарушения работы ассоциации эндотелиоцит-глиальная клетка (астроцит)-нейрон (»нейроваскулярное звено«) страдают механизмы ауторегуляции мозгового кровотока на уровне капилляров и микроартериол. Это приводит к сужению диапазона допустимых показателей перфузии. Из-за того, что мелкие сосуды утрачивают способность расширяться, становится невозможным перераспределение кровотока в пользу активно работающих отделов мозга, а это в свою очередь приводит к их функциональной инактивации, а затем – и к необратимому повреждению. Преимущественное поражение белого вещества в перивентрикулярном и глубинных отделах при церебральной гипоперфузии объясняется особым характером их кровоснабжения сосудами терминального типа, не имеющими коллатералей.
Клетки нервной ткани различаются по чувствительности к ишемии. При нарушении кровотока, некротические изменения развиваются сначала в нейронах, затем олигодендроцитах, миелинизированных аксонах, астроцитах и наконец в эндотелиальных клетках. После гибели наиболее чувствительных типов клеток — нейронов и олигодендроцитов, относительно резистентные к ишемии астроциты, выполняющие в головном мозге опорную функцию, замещают дефект за счёт увеличения в объёме своих отростков. Формируется глиальный рубец (»неполный» инфаркт»). При тотальной гибели всех элементов участка мозговой ткани формируется участок некроза (лакуна), на периферии регенерация глиальных клеток формирует его «оболочку».
В патологоанатомических исследованиях отмечена связь липогиалиноза мелких артерий с лакунарными инфарктами головного мозга. Указывается на более высокую распространённость расширенных периваскулярных пространств у пациентов с сосудистой деменцией, чем у пациентов с болезнью Альцгеймера. У здоровых пожилых пациентов с выявленными расширенными периваскулярными пространствами отмечались худшие результаты тестирования когнитивных функций.
Микроатероматоз — атеросклеротическое поражение артериол, приводит к развитию более крупных лакунарных инфарктов. В зависимости от локализации, инфаркты могут проявляться клиникой инсульта, но чаще (примерно в 80 % случаев) протекают субклинически (»немые» инфаркты). У части пациентов основные патологические процессы вызываются церебральной амилоидной ангиопатией (ЦАА) — отложением амилоидных пептидов (прежде всего Аβ — амилоида) в мышечной оболочке и адвентиции микрососудов и сосудов среднего калибра, значительно реже в стенках капилляров и вен.
Рисунок 4
Патогенез сосудистого повреждения головного мозга
Yang T, Sun Y, Lu Z, Leak RK, Zhang F. The impact of cerebrovascular aging on vascular cognitive impairment and dementia. Ageing Res Rev. 2016; 34:15–29.
Церебральная амилоидная ангиопатия в сочетании с НКР различных типов
Церебральная амилоидная ангиопатия (ЦАА) имеет сходные патогенетические механизмы с наиболее распространёнными типами НКР — сосудистым и БА. С первой группой состояний ЦАА роднит единство факторов риска и клинических проявлений, со второй — сходные патогенетические механизмы — формирование и накопление амилоидных β — пептидов. У пациентов с ЦАА отмечается повышение уровня общего и фосфорилированного тау-пептида в спинномозговой жидкости по сравнению с контрольной группой, менее значительное, чем при БА. Степень повышения уровня тау-пептида соответствует выраженности сопутствующей патологии БА.
В клинико-патологических исследованиях была показана связь умеренных и выраженных патологических проявлений ЦАА с когнитивным снижением. В двух недавних исследованиях, проводивших оценку профиля когнитивных нарушений пациентов с ЦАА (включались пациенты стационара, перенесшие развитие внутримозгового кровоизлияния без последующего развития деменции), были получены сходные результаты: в группах была отмечена большая доля пациентов с УКР, наиболее распространённым нарушением было снижение скорости обработки информации (Case NF, Charlton A, 2016; Harrison J K, Fearon P, 2014). Когнитивный профиль пациентов отличался от профиля, установленного в сходных популяционных исследованиях отсутствием выраженных нарушений памяти — основы клиники БА. Доказана связь ЦАА (вне зависимости от наличия сопутствующих кровоизлияний и патологии по типу болезни Альцгеймера) с последующим развитием деменции, клинические характеристики деменции сходны с сосудистой деменцией. Описана возможность первой манифестации ЦАА в виде других нейропсихиатрических нарушений: делирий, депрессия, расстройства личности.
.jpg)
Болезнь Паркинсона и болезнь диффузных телец Леви
Болезнь Паркинсона (БП) и болезнь диффузных телец Леви (БДТЛ) относятся к группе синуклеопатия (включающую также нейродегенеративное заболевание без когнитивных нарушений — мультисистемную атрофию).
Патологическая основа заболеваний включает формирование телец Леви — фибриллярных скоплений, состоящих из убиквитинированных белков нейрофиламентов и связанных с ними белков, которые накапливаются в результате повреждения клеток. Патологические скопления способствуют изменению структуры белков, нарушениям фосфорилирования и протеолиза (Pollanen М. S. с соавт. 1993). В настоящее время в составе телец Леви обнаружено свыше 90 компонентов. Убиквитин является обязательным компонентом всех телец Леви в коре мозга и свыше 90 % телец Леви ствола мозга (Fukuda Т. с соавт. 1993; Gai W. P. с соавт. 1996). Основным составным элементом телец Леви признан α — синуклеин (Wakabayashi К. с соавт. 2013).
Сравнительно небольшие агрегаты α — синуклеина обладают большей нейротоксичностью, чем более крупные — тельца Леви.
В основе БП и БДТЛ на молекулярном уровне лежит один и тот же патологический процесс, различия заключаются в последовательности вовлечения отдельных участков головного мозга. При БП развитие происходит «снизу вверх», от каудальных стволовых структур, вовлекая компактную часть чёрного вещества среднего мозга, подкорковые ядра и таламус и, в последнюю очередь, кору головного мозга (Braak с соавт. 2004). При развитии ДТЛ эта стадийность нарушается, патологический процесс может параллельно вовлекать кору головного мозга и периферические структуры вегетативной нервной системы, что объясняет преобладание в клинике болезни вегетативных и когнитивных нарушений над двигательными. Скорость развития патологических нарушений при ДТЛ выше, чем при БП. В финальной стадии, с распространённым вовлечением нервной системы, заболевания сходны как клинически, так и патогистологически.
Лобно-височная дегенерация (болезнь Пика)
Характерным признаком заболевания являются тельца Пика — аргирофильные глобулярные цитоплазматические включения, размер которых сопоставим с размером клеточного ядра (Bigio Е. Н. 2013). Они встречаются в нейронах неокортекса во II —III и VI слоях, а также в зернистых клетках зубчатой извилины, в пирамидных клетках гиппокампа и энторинальной коры и в некоторых подкорковых ядрах (Hof Р. К. с соавт, 1994) и часто образуют небольшие скопления (. Armstrong R. A. с соавт, 1998).
Тельца Пика состоят из прямых филаментов диаметром 15 нм, которые перемежаются более толстыми витыми филаментами с определенной периодичностью (Murayama S. с соавт, 1990). Иммуноцитохимические исследования показали, что основным компонентом телец Пика является гиперфосфорилированный белок тау, что относит болезнь Пика (лобно-височную дегенерацию) к таупатиям (Mori F. с соавт, 2002; Murayama S. с соавт, 1990; Probst A. с соавт, 1996). Однако позднее было обнаружено, что примерно половина случаев заболевания является таупатией с окраской телец Пика на белок ray, а в остальных случаях тельца Пика иммунонегативны на белок тау, но иммунореактивны на белок TDP-43 (transactive response DMA binding protein 43 к Da, TAR DMA-binding protein 43) (Bigio E. H. 2013). Таким образом, иммуногистохимический анализ позволяет выявить два подвида лобно-височной дегенерации: «таупатичсскую» и «TDP-43-патическую». При таупатической лобно-височной дегенерации в тельцах Пика наблюдается белок — FIG4 фосфатаза, участвующий в модификации фосфатидилинозитол-З, 5-бифосфата — молекулы, необходимой для внутриклеточного транспорта микропузырьков по эндосомо-лизосомальному пути. Однако FIG4 отсутствует при TDP-43-патической разновидности болезни (Коn Т. с соавт. 2014). В некоторых тельцах Пика обнаружен убиквитин (Bigio Е. Н. 2013).
Таупатическая форма является более распространенной. К «TDP-43-патической» форме относится существенная часть случаев семантического варианта первичной прогрессирующей афазии. Большинство случаев логопенического варианта ППА связаны с сопутствующими изменениями по типу БА, однако, от типичной формы болезни они отличаются асимметричным распределением нейрофибриллярных трубочек в больших полушариях головного мозга.
Болезнь Гентингтона
Болезнь Гентингтона (БГ) — врождённое прогрессирующее нейродегенеративное заболевание, характеризующееся хореиформными движениями, психическими нарушениями и деменцией. Развитие болезни связано с увеличением количества повторов кодона, кодирующего глутамин — цитозин-аденин-гуанин (ЦАГ) в участке 4p16.3 хромосомы 4; мутация приводит к удлинению полиглутаматного тракта в белке — продукте гена, который называется гентингтин. Гентингтин связан с клеточной мембраной и может оказывать влияние на везикулярный транспорт, синаптическую передачу и регуляцию аутофагии. Окончательное суждение о биологической роли гентингтина не сформировано.
Количество повторов ЦАГ коррелирует с возрастом начала заболевания, скоростью его прогрессирования и продолжительностью жизни пациентов.
Таблица 1
Классификация нуклеотидной последовательности и статус болезни, который зависит от числа кодонов ЦАГ (https://ru.wikipedia.org/wiki/Гентингтин)
| Число кодонов CAG | Статус выраженности мутации | Выраженность заболевания |
|---|---|---|
|
<28 |
Норма |
Норма |
|
28–35 |
Промежуточный |
Норма, при передаче возможно увеличение числа повторов. |
|
36–39 |
Частичная пенетрантность |
Шанс возникновения патологии относительно мал. |
|
40 и выше |
Полная пенетрантность |
Вероятность развития болезни — 100%. |
Для болезни характерен аутосомно-доминантный тип наследования.
У пациентов, страдающих заболеванием, в нейронах многих областей мозга выявляются внутриядерные включения. Эти включения иммунореактивны на гентингтин, убиквитин, р62 (Gourfinkel-An I. с соавт. 1998; Riib U. с соавт. 2014; Valera A. G. с соавт. 2005). Поскольку при этом заболевании наблюдается аномальный синтез полиглутаминовых последовательностей, при изучении различных отделов мозга используют антитела клона 1С2, позволяющие обнаружить полиглутамин. Оказалось, что 1С2-иммунореактивность присутствует во внутриядерных включениях нейронов ствола мозга (Rub U. с соавт. 2014), но не в коре и полосатом теле, где, однако, выявлялись цитоплазматические включения, иммунореактивные на 1С2 (Sieradzan К. A. [et al. ], 1999). Обнаружены также убиквитин — р62 — и 1С2-иммунореактивные включения в аксонах всех проводящих путей ствола мозга (Rub U. с соавт. 2014).
Общим патогенетическим звеном всех полиглутаминовых заболеваний является изменение конформации белков — частичное преобразование α — спирали в β — складчатость. Эти белковые цепи связываются в высокомолекулярные антипараллельные структуры по типу застёжки молнии, которые являются основой для амилоидных комплексов в клетках. Полиглутаминовая агрегация в нейронах запускается посттрансляционными протеолитическими изменениями гентингтина с N-концов при помощи каспаз (сериновых протеаз), кальпаина (кальцийчувствительная протеаза) и других эндопротеаз; полиглутаминовые фрагменты в усечённых фрагментах N-концов подвергаются воздействию окружающих субстратов и поэтому максимально «агрессивны».
Нейродегенеративный каскад БГ начинается после перемещения N-терминального фрагмента в ядро нейрона. Расщепление полиглутаминовых белков усиливает их способность проникать в ядро клетки, поскольку такой способностью обладают пептиды молекулярной массой <46 кДА.
Установлена роль митохондриальной дисфункции с нарушением энергетического обмена и нарушения работы цитоскелета клетки в лабораторных моделях БГ.
Эпидемиология
В развитых странах деменция является четвёртой по значимости причиной смерти.
Основным фактором риска развития деменции является возраст (Li R, Wang TJ, Lyu PY, 2018).
Серьёзный рост распространённости деменции с возрастом и снижение качества жизни пациентов и ухаживающих лиц в связи с этим, заметно уменьшают значимость увеличения продолжительности жизни в развитых странах.
Наиболее распространённым скрининговым инструментом является Краткая шкала оценки психического статуса (MMSE), однако, есть разногласия в вопросе выбора точки разделения (18,21,22,24 или 26/30 баллов), а также необходимости учёта возраста и уровня образования пациента.
В многоцентровом исследовании EURODEM, объединившем усилия всех крупных коллективов Западной Европы, было показано ступенеобразное увеличение распространённости деменции с возрастом пациентов — с 65–69 ти летнего возраста, каждые 5 лет доля лиц с деменций увеличивалась почти вдвое. Значимых половых различий не выявлено, однако, в возрасте до 75 лет среди пациентов с деменций была выше доля женщин, а после 75 лет — мужчин. Эти тенденции отмечались во всех 12 исследованиях, результаты которых использовались для анализа.
Также сходной была частота возникновения новых случаев заболевания: в возрасте 65 лет — 2.5 (95% ДИ 1,6 — 4,1), в возрасте 90+ — 85,6 (95% ДИ 70,4 — 104,0) на 1000 пациенто-лет. Значительное повышение риска возникновения деменции было связано с курением и низким уровнем образования. Травма с потерей сознания в анамнезе, женский пол, случаи деменции в семейном анамнезе не оказывали значительного влияния на риск развития деменции.
Работа группы, исследующей деменцию 10/66 проводилась в развивающихся странах (Индия, Китай, Нигерия, Куба, Доминиканская республика, Бразилия, Венесуэла, Мексика, Перу и Аргентина). Общее количество участников, закончивших исследование распространённости, составило 14960 человек. Распространённость деменции находилась в пределах 5,6–11,7%. Отмечены выраженные различия, зависящие от региона и выбранного места оценки.
К настоящему времени проведено несколько крупных популяционных долгосрочных исследований (Dening T. Thomas A. 2013).
Рисунок 5
Распространённость состояний, ассоциированных с возрастом. В отношении деменции показано самое большое повышение количества случаев с увеличением возраста пациентов (по данным исследования EURODEM)
Факторы риска
Основным фактором риска развития деменции является возраст. Роль средовых факторов и факторов риска сердечно-сосудистых осложнений отмечается в отношении всех типов НКР, однако, более выражено в пожилом возрасте. Учитывая, что наиболее распространённым типом НКР, является болезнь Альцгеймера, за основу при подготовке раздела взят набор факторов риска этого заболевания. В отношении ряда видом деменций (алкогольная, связанная с употреблением наркотиков, прионным заболеванием, перенесёнными инсультом или тяжёлой ЗЧМТ), действие причинного фактора настолько очевидно и сильно, что учёт других факторов риска не имеет практического смысла.
Генотип ApoE ε4 и другие генетические факторы риска
Наряду с возрастом, наиболее изученными факторами риска БА являются случаи деменции в семейном анамнез, редкие мутации в генах с доминантными типом наследования, оказывающих влияние на накопление амилоида в головном мозге, и ε4-аллель аполипопротеина Е (ApoE).
Роль генетических факторов выше в случаях БА с ранним началом, даже, в так называемых спорадических случаях. Развитие заболевания связано с аутосомно доминантным наследованием мутаций генов, оказывающих влияние на обмен белка-предшественника амилоида (APP), пресенилина 1 (PSEN1, до половины от всех случаев БА с ранним началом) и пресенилина 2 (PSEN2). Данные набор генетических аномалий отвечает за развитие примерно ⅔ случаев БА с ранним началом. Эти мутации имеют высокую пенетрантность (долю лиц-носителей гена, имеющих его фенотипические проявления), с вероятностью развития болезни в течение жизни, стремящейся к 100%.
Генетическая природа БА с поздним началом (сенильной формы) более сложная. Развитие заболевания связывается набором генетических факторов с меньшей пенетрантностью, предполагается большее влияние средовых факторов и факторов риска цереброваскулярных заболеваний (Caselli RJ, Dueck AC, 2011). Наиболее значимым генетическим фактором риска сенильной БА является фенотип ApoE. У пациентов-носителей ApoE ε4 риск развития БА выше в 8–12 баллов. В геномных исследованиях было выделено большое количество генов-кандидатов, однако, в большинстве случаев, их вклад в риск развития БА ограничивался 1,1–1,5 кратным увеличением.
Предполагается повышение риска развития сосудистой деменции у лиц-носителей ApoE ε4 фенотипа, однако в значительно меньшей степени, чем в отношении БА.
Нет единого мнения о влиянии фенотипа ApoE ε4 на риск развития деменции после полученной ЧМТ.
В популяционном исследовании, выполненном в Северном Манхеттене (Нью-Йорк), сочетание ЧМТ с наличием в генотипе пациента аллеля ApoE ε4 сочеталось с 10-ти кратным повышением риска деменции, в то время, как в отсутствии ApoE ε4, перенесённая ЧМТ повышала риск деменции лишь вдвое. В этом исследовании не было показано повышения риска деменции при перенесённой ЗЧМТ в отсутствии ApoE ε4. Сходные результаты были получены в проспективном исследовании ветеранов Второй мировой войны также показана большая вероятность деменции у носителей ApoE ε4. В исследовании MIRAGE, напротив, был выше у пациентов без аллеля ApoE ε4 (ОШ, 3,3), чем у гетерозиготных носителей ApoE ε4 (ОШ, 1,8) или гомозиготных носителей (ОШ, 1,3).
Цереброваскулярные и сердечно-сосудистые заболевания и их факторы риска
Артериальная гипертония
Артериальная гипертония в среднем возрасте сочетается с повышенным риском развития деменции и БА. Риск, вероятно опосредован развитием цереброваскулярного заболевания и других осложнений гипертонии. Лечение гипертонии снижало риск развития деменции только по данным обсервационных исследований, но не подтверждается в клинических исследованиях антигипертензивных препаратов.
Дислипидемия
В ряде исследований показана связь уровня холестерина липопротеинов низкой плотности (ЛПНП), особенно в среднем возрасте и риска БА. Однако, интерпретация этих результатов сложна, поскольку ЛПНП не проникают через интактный гематоэнцефалический барьер; большая часть холестерина в головном мозге синтезируется астроцитами и нейронами и поступает в нервные клетки в виде комплексов липопротеинов высокой плотности (ЛПВП).
Предполагается, что холестерин в веществе головного мозга может повышать риск БА, усиливая образование и/или отложение β — амилоида, а также оказывая влияние на другие факторы — риск цереброваскулярных заболеваний, местное воспаление и метаболизм тау-белка.
Липостатическая терапия представляет интерес в плане профилактики БА, однако, в рандомизированных исследованиях с участием пациентов с сердечно-сосудистыми заболеваниями не было показано протективного влияния статинов на когнитивные функции (однако, в исследованиях деменция не оценивалась как первичная конечная точка). Приём статинов также не влияет на вероятность конверсии лёгких форм БА в более тяжёлые.
Сахарный диабет и ожирение
Ожирение и сахарный диабет 2 типа повышают риск развития БА приблизительно в 1,5 раза. Механизм влияния этих состояний на развитие деменции окончательно не установлен. Предполагается изменение метаболизма амилоидов, в ряде исследований предполагается роль фермента, разрушающего инсулин (IDE). В ряде исследований показана связь патогенеза БА с уровнем инсулина в головном мозге.
Снижение физической активности
У физически активных лиц меньший риск развития и распространённость когнитивных нарушений, в том числе деменции, включая БА. В мета-анализе 16 проспективных исследований, после коррекции влияния других характеристик пациентов, было показано 28% снижение риска деменции в целом и 45% снижение риска деменции при БА
Перенесённые ЗЧМТ
Перенесённые черепно-мозговые травмы повышают риск развития деменции в 2–4 раза (Shively S, Scher AI, 2012).
Приём лекарственных препаратов
Бензодиазепины
Доступные данные о связи приёма бензодиазепинов и риска деменции противоречивы. По данным Канадского исследования с участием около 2000 пациентов (Billioti de Gage S, Moride Y, 2014), после коррекции других свойств пациентов (распространённость тревоги, депрессии, нарушений сна), приём бензодиазепинов в течение >180 дней сопровождался 1,5-кратным повышением риска развития БА. Риск находился в зависимости от длительности приёма препаратов и был выше у препаратов с большим временем полувыведения. Оценку полученных результатов нарушает возможность большей потребности в назначении бензодиазепинов у пациентов с продромальными симптомами деменции. В других крупных обсервационных исследования не было показано значимой связи длительного приёма бензодиазепинов с риском развития деменции (Koyama A, Steinman M, 2013,2014).
Антихолинергические препараты
В проспективном популяционном исследовании с участием > 3400 пожилых лиц без деменции на момент включения в исследование, которые наблюдались в среднем в течение 7 лет, приём антихолинергических в высокой накопленной дозе сочетался с повышением риска развития деменции (скорректированное отношение рисков 1,54 для наиболее высокой стандартизированной 95% ДИ 1,21–1,96). Наиболее распространёнными антихолинергическими препаратами были трициклические антидепрессанты, антигистаминные препараты первого поколения и М-холинолитики, использующиеся для лечения урологических проблем (Gray SL, Anderson ML, 2015).
Ингибиторы протонной помпы
Данные о связи приёма ингибиторов протонной помпы (ИПП) с риском развития деменции противоречивы. Результаты не менее чем двух исследований указывают существенное повышение риска развития деменции при приёме ИПП (Haenisch B, von Holt K, Wiese B, Prokein J, Lange C, 2015; Gomm W, von Holt K, 2016).
В одном проспективном когортном исследовании с участием 73000 лиц в возрасте 75 лет и старше, у которых исходно не было деменции, было показано, что регулярный приём ИПП сочетается с повышенным риском развития деменции (отношение рисков (ОР) 1,44,95% ДИ 1,36–1,52), связь сохранялось после удаления влияния множественных факторов, которые могут оказывать влияние, включая возраст, пол, наличие депрессии, перенесённого инсульта, заболеваний сердца и одновременный приём нескольких лекарственных препаратов (Gomm W, von Holt K, 2016). В менее крупном исследовании показана сходная связь риска деменции всех типов (ОР 1,3) и деменции Альцгеймеровского типа (ОР 1,4). Напротив, в двух других обсервационных исследованиях, включая большое исследование случай-контроль с участием 70000 пациентов с БА и исследование с участием более 280000 лиц, в котором характеристики основной и контрольной группы не отличались по поло-возрастным признакам и месту проживания, не было выявлено связи приёма ИПП с развитием БА (Taipale H, Tolppanen AM, 2017; Goldstein FC, Steenland K, 2017). В качестве возможного механизма влияния ИПП рассматривается нарушение всасывания витамина В12 и других питательных веществ. Не исключается наличие факторов, которые одновременно приводят к развитию деменции и необходимости приёма ИПП, для оценки этой гипотезы необходимы дополнительные исследования (Kuller L. H. 2016).
Средовые факторы риска
К средовым факторам риска деменции относятся факторы окружающей среды: действие пестицидов, тяжёлых (мышьяк, свинец — незначительное повышение риска) и других металлов (алюминий, цинк) загрязнение окружающей среды, пассивное курение (Killin LO, Starr JM, 2016).
Влияние деменции на ожидаемую продолжительность жизни
По данным Роттердамского исследования (10348), которое проводилось с 1990 по 2015 годы и фиксировало случаи развития деменции и смерти,
Ожидаемая продолжительность жизни женщин находилась в пределах от 18,0 лет (95% ДИ 17,8–18,2), если диагноз «деменция» был установлен в возрасте 65 лет, до 2,3 лет (2,2–2,3) при постановке диагноза в 95-ти летнем возрасте; у мужчин 15,6 (15,4–15,9) и 1,8 (1,7–1,8). Увеличение возраста развития деменции на 1–3 года приводило к уменьшению продолжительности жизни с диагнозом деменция на 25–57% (Wolters FJ, Tinga LM, 2018).